genellik
Prader-Willi sendromu fiziksel, davranışsal ve entelektüel anormalliklere neden olan nadir bir genetik hastalıktır. En karakteristik klinik belirtiler obezite (ve ilişkili hastalıklar) ve düşük kas tonusudur.
Objektif muayene genellikle doğru tanı koymak için yeterlidir, ancak güvenilir genetik testler yapmak da mümkündür.
Ne yazık ki, henüz bir çözünürlük tedavisi yoktur; ancak, bazı farmakolojik ve davranışsal karşı önlemler ilişkili semptomatolojiyi sınırlayabilir.
Genetiğe kısa referans
Prader-Willi sendromunu tarif etmeden önce, genetiğe kısa bir referans yapmak iyidir.
KROMOSOMLAR VE DNA
Sağlıklı bir insanın her hücresinde 23 çift homolog kromozom vardır : 23'ü annedir, yani anneden, 23'ü babadan veya babadan kalıtılır. Bu kromozomların birkaçı cinseldir, yani bireyin cinsiyetini belirler; kalan 22 çift ise otozomal kromozomlardan oluşmaktadır. Bütünlüklerinde, 46 insan kromozomu, DNA olarak bilinen tüm genetik materyali içerir. Bir bireyin DNA'sında somatik özellikleri, eğilimleri, fiziksel armağanları vb. Yazılır.
GENLER VE DNA MUTASYONLARI
DNA, gen adı verilen az çok uzun dizilerde düzenlenir.

Şekil: Bir genin bir çift homolog kromozom içinde organizasyonu. Bir çift homolog kromozom, hepsi iki değişkene sahip allelleri olan, aynı kromozomal pozisyonda olan ve aynı fonksiyonları gerçekleştiren (mutasyonlar hariç) spesifik genler içerir. Soldaki kromozom çiftinin iki eşit aleli vardır (her ikisi de açık mavi); Öte yandan, sağ çift iki farklı alele sahiptir (biri kırmızı, diğeri mavidir).
Her bir gen, belirli bir kromozomu ve homologunu işgal eder, çünkü iki kopyada var olduğu gibi allel denir. Allel anneden gelir ve maternal kromozomda bulunur; diğer alel babadan gelir ve baba kromozomunda bulunur.
Genlerden, vücudumuzda bulunan proteinleri çıkarır. Bir DNA mutasyonu gerçekleştiğinde, belirli bir kromozomun bir geni (genellikle bir allel) ya kusurlu olabilir ve hatalı bir protein üretebilir ya da tamamen başarısız olabilir.
Prader-Willi sendromu nedir?
Prader-Willi sendromu, 15 kromozomunun değişmesine bağlı bir dizi fiziksel, entelektüel ve davranışsal bozuklukla karakterize nadir bir genetik hastalıktır.
En erken çocukluk döneminde, sendrom olağandışı kas zayıflığı ve gelişimsel bir gecikme ile kendini gösterir. Daha sonra çocukluk döneminde iştahsızlık, öğrenme güçlüğü ve davranışsal anormallikler gibi başka sorunlar da ortaya çıkmaya başlar.
Prader-Willi sendromlu bireyler, sıklıkla, çeşitli şekillerde kalp problemlerinden kaynaklanan, obezite muzdarip insanlardır. Sonuncusu ölümün ana nedenidir.
epidemioloji
Prader-Willi sendromu nadir görülen bir durumdur: aslında, etkilenen bir çocuğun doğumu her 15.000 - 30.000 yenidoğanda ortalama olarak kaydedilir.
Erkekleri ve kadınları eşit ölçüde etkiler ve belirli ırklar için ön tercihi yoktur.
Nedenler
Prader-Willi sendromuna neden olan neden, kromozom 15 düzeyinde genetik bir mutasyondur . Kesin etkilenen gen henüz netleştirilmemiştir; şüpheliler, her şeyden çok, birden fazla gen içeren kromozomal bir bölgeye düşer.

Şekil: Bir kromozom 15 ve Prader-Willi sendromunda rol oynadığından şüphelenilen genler. Web sitesinden: www.kreatech.com
KROMOSOM'TA GENLER 15
Genellikle, vücut hücrelerimiz protein oluşturmak için her iki aleli kullanır. Başka bir deyişle, bu hem kromozomların hem anne hem de babaların yararlı olduğu ve genetik katkılarını sağladığı anlamına gelir.
Bununla birlikte, bazı özel hücrelerde, evrimsel ve patolojik olmayan bir mesele için, sadece bir (baba veya anne) aleli çalışır ve işi tatmin edici olmaktan iyidir. Aktif olmayan alel, tam olarak var olduğu, ancak kendisini "ifade etmediği" için sessiz olarak adlandırılır.
Beynimizin hücrelerinin tümü kromozomu 15 içerir, ancak bazı bölgelerde sadece maternal genetik çizgi ifade edilirken, bazılarında sadece baba çizgisi ifade edilir. Prader-Willi sendromundan sorumlu beyin bölgesi olan hipotalamusta, sadece paternal kromozom genleri normalde eksprese edilir.
KROMOSOM 15 VE YAŞAYICI-WILLI SENDROMU
Genetik testlerle Prader-Willi sendromlu hastaların normal bir 15 kromozomu bulunmadığı tespit edildi. Bu sadece aktif kromozomal çizginin baba olan olduğu hipotalamusta zararlıdır.
Hipotalamusun ana fonksiyonları:
- İştah düzenleme
- Uyku-uyanma ritimlerinin ayarlanması
- Duygusal durumların ifadesi
- Vücut ısısının düzenlenmesi
- Hormon üretimi
Fakat baba kromozomu 15'in arızasını veya yokluğunu tetikleyen nedir? Muhtemel sebepler en az üç:
- Belirli bir paternal kromozom bölgesi 15 yokluğu: aslında, kromozomun önemli bir kısmı yoktur.
- İki maternal 15 kromozom. Bu anomali embriyo oluşumu sırasındaki bir hata nedeniyle oluşur.
- Paternal kromozom 15 üzerinde mevcut bazı genlerin değişimi.
GENETİK VE MİRAS VEYA SADECE GENETİK?
Genetikçiler Prader-Willi'nin sendromunu genetik bir hastalık olarak kabul ederler, çünkü yapılan çalışmalara göre, yukarıda belirtilen üç mutasyon yönteminin ebeveynlerden miras alınmadığı (normal bir kromozomal kitine sahip olan) ancak tesadüfen ortaya çıktığı ortaya çıkmıştır. gebe kalmadan hemen önce ( sporadik mutasyon ).
Bununla birlikte, bu bildirimin doğruluğu, Prader-Willi sendromu olan birden fazla oğlu olan bazı çiftlerin ebeveynlerinin tespiti ile zayıflatılmıştır. Bu gibi durumlarda, hastalığın kökeninde hala kanıtlanmış, kalıtsal bir bileşenin olabileceğine inanmak için sebep vardır.
PRADER-WILLI SENDROMU VE ANGELMAN'IN SENDROMU
Prader-Willi'nin sendromu, bir şekilde, Angelman sendromunun zıttıdır: ikincisi, aslında, maternal kromozom 15 düzgün çalışmaz.
Belirtiler ve Komplikasyonlar
Derinleştirmek için: Prader-Willi sendromu belirtileri
Prader-Willi'nin sendromu, zaten ilk çocukluk döneminde (yaşamın ilk yılında), ilk semptom ve bulgularla kendini gösterir; Bu dönemde, esas olarak kas tonusunda (hipotoni) bir azalmaya ve gelişimde bir gecikmeye neden olur. Zamanla, hastalık daha sonra semptomatik tabloyu daha da zenginleştiren bir çeşit evrime doğru gidiyor.
ÇOCUKLUK
Yaşamın ilk yılında, ana işaretler şunlardır:
- Kas hipotoni . Bu kasların tonusunun normale göre azaldığı anlamına gelir: genellikle annenin sütün zor emilmesiyle olduğu kadar yumuşak uzuvlarda ve çok reaktif değil de ortaya çıkar.
- Gelişimsel gecikme . Hipotoniye bağlı olarak emme zorlukları tarafından tercih edilme eğilimindedir.
- Şaşılık .
- Karakteristik yüz özellikleri . Badem gözler, tapınaklarda başın daralması, ağzı aşağıya dönük ve ince üst dudak.
- Uyarıcılara yanıtın kısmen veya tamamen yokluğu . Çocuk yorgun ve onu uyandırmak kolay değil.
FANCIULLEZZA'DAN YAŞA? YETİŞKİN
Yaşamın ilk yılından sonra, dramatik bir sonucu olabilecek uzun bir dizi sorun ortaya çıkar.
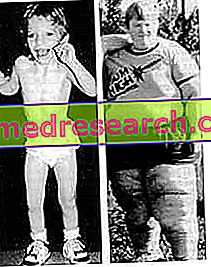
- Olağanüstü iştah ve obezite . Hastalar, yiyecek için sürekli bir istek gösterir, bu da onların çok yemek yemesine ve kilo almasına neden olur. Yiyecek bulamazlarsa, donmuş yiyecek ve atık tüketmeye başlarlar, yani yenilebilir bir şey. Bütün bunlar hipotalamusun değişen fonksiyonlarından kaynaklanmaktadır.
- Hipogonadizm . Bu, genital organların (testisler, erkeklerde ve kadınlarda, kadınlarda) az sayıda seks hormonu (erkek testosteron ve kadın östrojen) ürettiği anlamına gelir. Hasta ergenlik gelişimini tamamlamaz ve genellikle verimli değildir. İlk adet, kadınlarda ertelenir (eğer tamamen yoksa); erkekte seste bir değişiklik gözlenmez.
- Büyüme ve azaltılmış gelişme . Kalan kas hipotonisi problemine, ergenlik döneminden sonra bile (genellikle ergenlerin birkaç santimetre yükseldiği) düşük bir statü gelişimi ekleriz.
- Öğrenme açığı . Hastaların entelektüel fakülteleri neredeyse her zaman azalır.
- Davranış problemleri . Özellikle ergenlik döneminde, bireyler inatçı, kaprislidir ve obsesif-kompulsif bozukluğa maruz kalırlar.
- Motor gecikmesi Çocuklar çok geç yürümeyi öğrenirler.
- Dilin zorluğu . Genellikle hastalar ciddi bir gecikmeyle konuşmaya başlar ve dilleri zayıf ve zor kalır.
- Uyku bozuklukları REM ve REM OLMAYAN uyku fazları arasındaki normal değişime uyulmaz. Ek olarak, hastalar uyurken, solunum kesintilerinden muzdariptir (uyku apnesi).
- Skolyoz . Sorun yalnızca bazı hastalar için ayrılmıştır.
DOKTORA İLETİŞİM NE ZAMAN
bebekte. Prader-Willi sendromu şüphesine yol açması gereken belirtiler şunlardır: gelişme eksikliği, kas hipotoni, anne sütünü emmedeki zorluklar, yüz özellikleri ve uyaranlara cevap vermeme.
Çocukta İki temel ipucu: sürekli yiyecek arayışı ve ağırlıktaki hızlı artış.
KOMPLİKASYONLAR
Prader-Willi sendromunun ana komplikasyonları obezite ve diyabet, kalp hastalığı, hipertansiyon, hiperkolesterolemi, ateroskleroz, vb. İle ilişkili tüm sorunlardan kaynaklanmaktadır. Üstelik, sürekli beslenme bağlamında kalan, titizlikle tüketilen bir öğün nedeniyle hastanın boğulması kolaydır.
Çok önemli komplikasyonların bir diğer serisi hipogonadizm ile bağlantılıdır: cinsiyet hormonlarının eksikliği çoğu zaman kısırlığa ve osteoporoza neden olur.
tanı
Genetik testleri kullanmadan önce, basit bir objektif inceleme ve bazı kan testleri ile Prader-Willi sendromunun doğru bir ön teşhisi konulabilir.
Fizik muayenede bulunabilecek klinik bulgular
bebekte:
- Kas hipotoni
- Badem gözler
- Tapınakların Tapınakları
Çocukta / ergende:
- Doyumsuz iştah
- şişmanlık
- Davranış problemleri
Genetik testler doğrulama görevi görür ve hastalığa neden olan mutasyon tipinin netleşmesine yardımcı olur.
tedavi
Ne yazık ki, bu genetik bir hastalık olduğundan, Prader-Willi'nin sendromu tedavi edilemez.
Uygulanabilir tek terapötik tedaviler semptomları sınırlandırmayı (örneğin obeziteyi), bazı anormal davranışları ılımlılaştırmayı ve genel olarak hastaların yaşam standartlarını iyileştirmeyi amaçlar.
Tüm bunları başarmak için, endokrinolojiden dietolojiye, fizyoterapiden psikoterapiye, farklı alanlarda uzmanlaşmış doktor ve uzmanlardan oluşan bir ekiple bağlantı kurmanız önerilir.
Aşağıda, en yaygın terapötik önlemler bildirilecektir.
GÜVENLİK VE SONRAKİ ADIMLARDA BESLENME
Erken çocukluk döneminde, emme ve kusurlu gelişme zorluklarını telafi etmek için, çocuğa yüksek kalorili yemekler vermek iyidir.
Aşağıdaki aşamalarda, durum tamamen değişmektedir: Uygulanan yemekler kalorilere azami dikkat gösterilerek dikkatlice kontrol edilmelidir.
Tavsiye istemek için en uygun uzman diyetisyendir .
BÜYÜME HORMONU
Ekzojen (yani dışarıdan) büyüme hormonu ( GH ) uygulamasının üç etkisi vardır:
- Aksi takdirde eksik olacak olan büyümeyi teşvik etmek
- Kas tonusunu iyileştirmek
- Vücut yağ seviyelerini azaltmak
Tedavi yaklaşık 3-5 yaşlarında başlar.
Günümüzde laboratuvarda oluşturulan ve yan etkileri azaltılmış hormon preparatları bulunmaktadır.
Bu durumda en uygun uzman endokrinologdur .
CİNSEL HORMONLAR
Testosteronun, erkekler için ve kadınlar için östrojenin ekzojen olarak verilmesi, bu iki hormonun azalan seviyelerini restore etmek için şarttır. Doğurganlığın arttırılmasına ek olarak, hormon tedavisinin osteoporoz üzerinde de etkileri vardır.
Tedavi ergenlikle başlar.
Daha fazla bilgi için: Prader-Willi Sendromu Tedavi İlaçları »
FİZYOTERAPİ VE LOGOPİ
Prader-Willi sendromlu hastalar fiziksel rehabilitasyon ve dil gerektirir. İlk kas hipotoni ve obezitenin etkilerini sınırlamayı amaçlamaktadır; sözlü ve yazılı iletişim eksiklikleri için ikinci çare.
Açılacak olan uzmanlar sırasıyla bir fizyoterapist ve bir konuşma terapistidir.
PSİKOTERAPİ VE MESLEKİ TEDAVİ
Psikoterapi, obsesif kompulsif bozuklukları ve genel olarak ruh hali olan hastalar için önemlidir. Bir psikiyatrın veya psikoloğun desteği, davranışsal yönü büyük ölçüde iyileştirebilir.
Mesleki terapi ise hastaya kendilerine nasıl bakacağını, nasıl giyineceğini vb. Başka bir deyişle ana günlük aktivitelerini nasıl gerçekleştireceğini öğretmeyi amaçlar.
AİLE'NİN YARDIMI
Ailenin yakınlığı, özellikle gençlik yıllarında hasta akrabasına yardım etmek için esastır. Genellikle ailelere verilen tavsiyeler, hastayı tüm faaliyetlerinde (özellikle beslendiğinde) takip etmek, dışlamak değil, onu ayırmak için en uygun davranışı araştırmaktır.
Prognoz ve korunma
Prader-Willi sendromu tedavi edilemez bir hastalık olduğundan prognoz pozitif olamaz. En büyük tehlike, obezite ve buna bağlı patolojiler tarafından temsil edilir: ölüm genellikle bunlardan birinden kaynaklanır.
Mevcut tedaviler (dengeli beslenme, hormon tedavisi, psikoterapi, vb.) Yaşam kalitesini hassas bir şekilde bile iyileştirir; Bununla birlikte, semptom kontrol önlemleri kalır ve daha fazlası olmaz.
Akrabaların yakınlığı esastır: destekleri aslında hastaların ömrünü uzatabilir.
ÖNLEME
Hastalık genetik bir mutasyon nedeniyle embriyoda ortaya çıktığında, onu önlemenin bir yolu yoktur.
Bununla birlikte, iki ebeveyn zaten Prader-Willi sendromlu bir çocuğu doğurduysa, ikinci bir hamilelikten önce, hastalığın taşıyıcı olup olmadığını öğrenmek için bazı genetik testlerden geçebilirler.