Marfan Sendromu Nedir?
Marfan sendromu, öncelikle gözleri, kardiyovasküler sistemi ve iskelet kası sistemini etkileyen karmaşık bir kalıtsal bağ dokusu bozukluğunu tanımlar. Bununla birlikte, her organın bağ dokusundan oluştuğunu göz önünde bulundurarak, Marfan sendromu ideal olarak her anatomik bölgenin büyümesini ve fonksiyonunu bozabilir ve engelleyebilir.
Sendrom, otozomal dominant bir özellik olarak bulaşır: bu nedenle oldukça değişken bir fenotipik ekspresyona sahip şiddetli genetik bir hastalıkla karşı karşıya kalıyoruz (kusurlar aileden aileye veya hastadan hastaya çok büyük farklılıklar gösterebilir).
Marfan sendromunu tetikleyen, mikrofibriller için yapısal destek olan konektörün çok önemli bir glikoproteini olan fibrilin-1'i kodlayan FBN1 geninin (kromozom 15 üzerinde) değiştirilmesidir.
Mikrofibriller: fibrilinden oluşan mikrofibiller, elastinin elastik liflerde birikmesi için bir interklasyonu oluşturduğu hücre dışı matriste bulunur. Her ne kadar vücutta omnipresent olsa da, mikrofiberler her şeyden önce aortta, bağ gövdeleri ve siliyer cisimlerin zonüllerinde bol miktarda bulunur (oküler seviye).
Otozomal dominant bir hastalık olarak, sadece her iki ebeveyn tarafından değiştirilen bir FBN-1 genini miras alan çocuklar Marfan sendromundan etkilenir. Bununla birlikte, 4 olguda bir vakada hastalık aile öyküsü olmayan hastalarda spontan mutasyonların sonucudur.
Hastalığın adı, ilk olarak 1896'da (A. Marfan) tekrar tanımlayan Fransız çocuk doktorundan geliyor, ardından semptomatolojik tezahürde yer alan değiştirilmiş geni tanımlamak için 1991 yılına kadar beklemek gerekliydi: keşif F. Ramirez idi.
Videoyu izle
X Youtube'daki videoyu izleyinNedenler
Marfan sendromunun fibrillin-1'i kodlayan bir genin mutasyonunun hemen ifadesi olduğunu belirtmiştik. Sendromu tetiklemek için hangi mekanizmanın kurulduğunu anlamak için tartışmayı derinleştirmeye çalışıyoruz.
FIBRILLINE 1, elastinin ve elastikiyetin ve doku mukavemetinin sağlanması ve sürdürülmesi için gerekli olan bir glikoprotein bileşenidir. Fizyolojik koşullar altında, fibrilin 1, TGF-beta (veya dönüştürücü büyüme faktörü beta) olarak bilinen başka bir proteine bağlanır. TGF-beta, vasküler düz kas ve hücre dışı matrisin zararlı işlemlerinde yer almaktadır. Bu varsayımlardan yola çıkarak, bazı yazarlar, Marfan sendromunun, FBN-1 geninin mutasyonuna ek olarak, ayrıca kalp kapaklarında ve akciğerlerde, özellikle aortta, TGF-beta fazlalığına bağlı olduğuna inanmaktadır.
oran
Marfan sendromunun her 3, 000-5, 000 doğumda bir 1 kişiyi etkilediği ve kesin bir yarış için önceden bir seçim yapmadan kendisini ve kadınlarını ayırt edici bir şekilde gösterdiği tahmin edilmektedir. İstatistikler, hastaların% 75'inin aile geçmişinin olumlu olduğunu göstermektedir; Kalan% 25'inde ise sebep, bir şekilde, hamile kalma anındaki ilerleyen yaşla ilişkili gibi görünen sporadik mutasyonlarda bulunur.
Marfan sendromunun aşırı şiddetli formları olan çocukların bir yıldan daha az bir yaşam beklentisi vardır.
Açık kalp ameliyatı stratejilerinin gelişmesinden önce, Marfan sendromlu hastaların çoğunda ortalama yaşam süresi 32 yıldı; Tıbbi ve farmakolojik tedavilerin sürekli iyileştirilmesi sayesinde, şu anda Marfan sendromu hastaları ortalama 60 yıla kadar yaşıyor.
İşaretler ve belirtiler
Derinleştirmek için: Marfan Sendromu Belirtileri
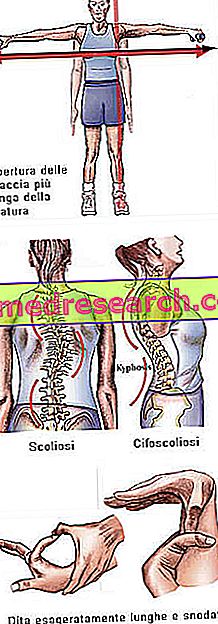
Marfan sendromu tamamen asemptomatik başlayabilir. Etkilenen hastalar abartılı bir şekilde uzun çizgi yapısına sahiptir, orantısız şekilde uzun ve incedir. Alt ve üst uzuvların gövdesi gövdeden (dolicostenomegali) çok daha yüksek bir uzunluğa sahiptir. Ayrıca, Marfan sendromundan etkilenenlerin tipik olarak abartılı parmak uzunluğu kavramını daha iyi ifade etmek için araknodaktik olarak da konuşuruz: bu nedenle eller bir örümceğin bacaklarıyla karşılaştırılır.
Boy ile ilgili olarak, bu hastalar yüzde 97'nin üzerinde bir ortalama ile kısa.
Marfan sendromu olan hastalarda sıklıkla görülen diğer ayırt edici özellikler arasında ayrıca şunu da belirtiyoruz:
- Yükseklikten daha büyük kolların açılması
- Yanal eklemler → abartılı eklem hareketliliği
- Göğüs duvarının deformitesi
- Lens çıkığı
- Üst vücut alt bölgeden daha az gelişmiştir
- Spontan pnömotoraks (% 11)
- skolyoz
- Uyluk, sırt, deltoid, pektoral seviyede cilt çizimleri
Marfan sendromuyla ilişkili en sorunlu bulgular arasında kalp kapak prolapsusu ve mitral kapak yetersizliğini hatırlıyoruz: benzer bir durum aortik halka genişlemesini ve aort diseksiyonunu kolayca lehimleyebilir.
Tablo, Marfan sendromu hastalarında bulunabilecek işaretleri göstermektedir. Açıklanan karakterler her zaman mevcut değildir, ancak bunların iyi bir kısmı bulunabilir.
| Etkilenen anatomik bölge | Olası semptomlar |
sevimli | Torasik, lomber ve sakral alanlarda stria |
gözler | Görme değişikliği, astigmatizma, retinanın ayrılması, açı kapanması glokomu, lens çıkığı, miyopi |
Kemik yapısı | Artralji, kifoskolyoz, dolikhostenomelia (gövde ile karşılaştırıldığında uzuvların aşırı uzunluk gelişimi), hipermobilite, yüksek damak, deforme göğüs, düz ayaklar, dar ve dar bilekler, anormal yeniden giriş / sternum çıkıntısı, skolyoz, kavisli omuzlar, spondilistezis |
Dita | araknodaktili |
akciğer | Spontan pnömotoraks, dispne, idiyopatik pulmoner obstrüktif hastalık |
Yüz değişiklikleri | Ogal damak (damakta malformasyon), mandibular retrogniite (çenenin gelişim bozukluğu), uzun yüz |
kalp | Anjina pektoris, abdominal aort anevrizması, kardiyak aritmi, torasik aort dilatasyonu / rüptürü / diseksiyonu, aort yetmezliği, mitral kapak prolapsusu |
dil | Dilin zorluğu |
tanı
200'den fazla olası mutasyon göz önüne alındığında, genetik belirteçlerin kullanımı teşhis amaçlı neredeyse imkansızdır.
Mutasyonun fenotipik ifadesinin tanımlanması her zaman açık ve basit olmadığı için Marfan sendromunun değerlendirilmesi her zaman bu kadar acil değildir. Tanısal gecikme hastanın sağkalımını ciddi şekilde tehlikeye atabilir: sadece bir kardiyovasküler problemin tanınmamasını düşünün.
Marfan sendromu için tanısal kriterler 1996 yılında uluslararası olarak hazırlanmıştır: tanı, sendromun majör ve minör göstergelerinin kombinasyonuyla ilişkili bir aile öyküsü anketinden oluşmaktadır.
Kullanılan birçok tanı testinden bazıları:
- ekokardiyogram
- manyetik açısal rezonans ve BT (aort incelemesi için)
- Kontrast sıvılı manyetik rezonans anjiyografi (MRA) (iç aort yapılarını görünür kılmak için)
- yarık lambalarla inceleme (lensin muhtemel yerinden çıkmasını analiz etmek için)
- oküler basınç ölçümü (olası glokom varlığını vurgulamak için)
- genetik testler (sendromu tespit edip etmemesi için çocuk düşünmeden önce önerilir)
terapiler
Genetik bir hastalık olduğundan, hastalığı tersine çevirebilecek bir ilaç ya da tedavi yoktur.
Bununla birlikte, semptomları hafifletmek ve herhangi bir komplikasyondan, özellikle de kalp krizinden kaçınmak için ilaçların kullanılması esastır. Bu amaçla, sartans (özellikle), ACE inhibitörleri ve beta blokerleri gibi kan basıncını düşüren ilaçlar özellikle belirtilmiştir.
Marfan sendromu bağlamında, skolyozdan muzdarip hastalar ayrıca glokomdan etkilenenlerin yanı sıra özel bakım da yapabilirler.
Cerrahi, Marfan sendromlu hastaların çoğunu birleştiren bir element olan anormal aort dilatasyonunu düzeltmek için düşünülebilir.
Devam: Marfan Sendromu - İlaçlar ve Bakım »